| [Site Map][Japanese][English] |
-
Student Recruitment
-
Project
- Computational Science
- Nonequilibrium Systems
- Chemical Reactions
- Macroscopic Chemical Phenomena
- Life Phenomena
- Multiscale Information Processing
- Industry Applications
- Meetings
- FY 2022
- FY 2021
- FY 2020
- FY 2019
- FY 2018
- FY 2017
- FY 2016
- FY 2015
- FY 2014
- FY 2013
- FY 2012
- FY 2011
- FY 2010
- FY 2009
- FY 2008
- FY 2007
- FY 2006
- FY 2005
- FY 2004
- FY 2003
- FY 2000
- FY 2019
- FY 2018
- FY 2017
- FY 2016
- FY 2015
- FY 2014
- FY 2013
- FY 2012
- FY 2011
- FY 2010
- FY 2009
- FY 2008
- FY 2007
- FY 2004-2006
- FY 2003
ConceptResearchesMeetings & Seminars
Open Seminars
References
Facilities
-
People
- Staff
- PostDocs & Students
- OB & OG
- OB & OG Messages
- FY 2018-
- FY 2015-2017
- FY 1999-2014
MembersSnapshots -
Access
-
Link

- Concept
- Computational Science
- Researches
- Nonequilibrium Systems
- Chemical Reactions
- Macroscopic Chemical Phenomena
- Life Phenomena
- Multiscale Information Processing
- Industry Applications
- Meetings & Seminars
- Meetings
- Open Seminars
- References
- Materials
- Publications
- Lecture Material
- Facilities
- Computers
From Isolated Reactions to Condensed phase Chemical Reactions
Nowadays, we can easily obtain such information as the geometric structures and potential energy surfaces in isolated reaction systems. On the other hand, in condensed phase chemical reaction systems such as solution, it is quite difficult to clearly define such microscopic features since not only solute molecules but also a vast number of the solvent molecules around them are related in the chemical reactions. Under this circumstance, we have proposed the "free energy gradient (FEG) method" as one of the theoretical methods to uniquely determine the equilibrium structure in solution and to clarify the solvation structures and dynamics of the solute molecules in condensed phase chemical reaction systems, and applied it to various ones.
Theoretical study on glycine isomerization reaction in aqueous solution
Glycine molecule is the smallest amino acid. Its zwitterion (ZW) does not exist stably since it is a nonionic molecule in gas phase. On the other hand, it is known that the neutral (NF) and ZW glycine molecules coexist in aqueous solution. With the FEG method, we have reproduced quantitatively the isomerization reaction in aqueous solution, and obtained the optimized structures in both stable and transition states. Figure 1 shows the free energy profile of the glycine isomerization reaction by the FEG method (horizontal axis is intrinsic reaction coordinate). In aqueous solution, the glycine molecule that exists in the NF (left balloon) in gas phase became stable as the ZW (right balloon), and the clear transition state (middle balloon) was observed between NF and ZW. In addition, the NF glycine was found to take eight stable conformers, and we have comprehensively investigated the relative stability between these conformers. As a result, it was shown that the free energy landscape in aqueous solution becomes rather flat in comparison with that in gas phase due to the offsets between the stabilization from the solute-solvent electrostatic interactions and the potential energy destabilization of the solute molecule itself.
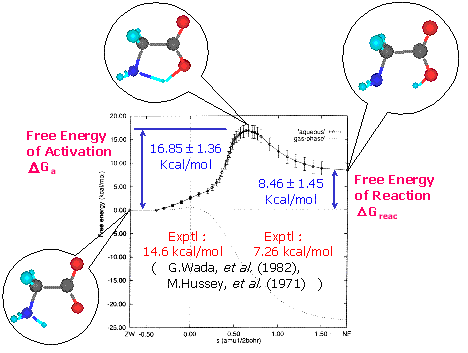
Figure 1. Energy profile of glycine isomerization reaction
[Reference]
N. Okuyama-Yoshida, K. Kataoka, M. Nagaoka and T. Yamabe, J. Chem. Phys., 113, 3519 (2000).
N. Takenaka, Y. Kitamura, Y. Koyano, T. Asada and M.Nagaoka, Theor. Chem. Acc., 130, 215 (2011).
Y. Kitamura, N. Takenaka, Y.Koyano and M. Nagaoka, Chem. Phys. Lett., 514, 261 (2011).
Theoretical study on ammonia ionization reaction in aqueous solution
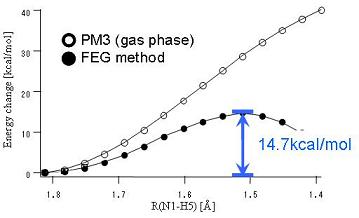
Fig.2 Energy profile of ammonia ionization reaction
Ammonia easily liquefies under high pressure and has high solubility in water. We have executed the solute geometrical optimizations by FEG method, and obtained the free energy profile of the ionization reaction in aqueous solution. As shown in Figure 2, the solute potential energy was destabilized in the gas phase (○) as its reaction proceeds and the transition state (TS) structure does not exist while a clear TS structure was seen on the reaction path in aqueous solution (●).
The calculated value of activation for ionization (14.7 kcal/mol) was found to be slightly over the experimental one (9.57 kcal/mol).
[Reference]
M. Nagaoka, Y. Nagae, Y. Koyano, Y. Oishi and M. Nagaoka, J. Phys. Chem. A, 110, 4555 (2006).
Y. Koyano, N. Takenaka, Y. Nakagawa, M. Nagaoka, Bull. Chem. Soc. Japan, 83, 486 (2010).
Development of theoretical methodology to interpret vibrational spectra
In addition to structural information (i.e., stable structures, intermediates and transition ones), the state of motion is very useful to understand the chemical reactions. The vibrational frequency analysis (VFA) has been implemented in the quantum chemistry calculation packages, such as GAUSSIAN, as a common theoretical technique, due to not only the comparison with the experimental vibrational spectrum but the properties of undergoing a chemical reaction, i.e., the equilibrium constant and reaction rate. However, the vibrational motions of the chemical species in condensed phase are complicated by interactions with the surrounding solvent molecule in comparison those in gas phase.
We have proposed the dual approach to vibrational spectra (dual VFA approach) as approximate methodology to effectively estimate how the molecular vibrates in condensed phase, and investigated the influence of microscopic solvent effects (such as the hydrogen bonding) on vibrational spectra. Figure 3 represents the IR spectra of a water molecule in liquid.
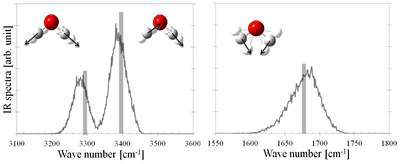
Figure 3. IR spectra of water molecules in liquid water
[Reference]
H. C. Georg, T. S. Fernandes, S. Canuto, N. Takenaka, Y. Kitamura, M. Nagaoka, In Practical Aspects of Computational Chemistry III; J. Leszczynski, M. K. Shukla, Eds.; Springer; New York, USA, 2014.
Y. Kitamura, N. Takenaka, Y. Koyano, M. Nagaoka, J. Chem. Theor. Comp., 10(8) 3369 (2014).
Theoretical study on associated states of methyllithium
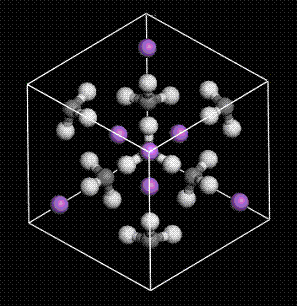
(gray: carbon, white: hydrogen, purple: lithium; space group: I-43m)
Alkyllithium (RLi) is an organometallic compound which plays a quite important role as a nucreophilic reagent in organic chemistry in synthesis because it is a strong base. The crystal of methyllithium (MeLi), which is one of RLis, exists as the body-centered cubic lattice of tetrameric aggregation (each methyl group forms the staggered conformation for each Li atom (see Fig. 4)).
On one hand, the structure of the isolated MeLi in gas phase has been interpreted variously by both theoretical and experimental approaches (in contrast to MeLi crystal, each methyl group forms the eclipsed conformation for each Li atom (see Fig. 5)).
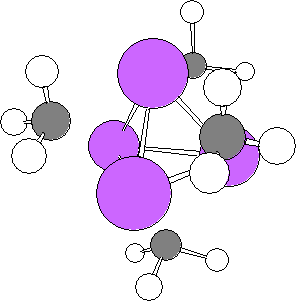
On the other hand, MeLi in organic solution exists as tetramer (in THF) or as the chemical equilibrium state between tetramer and dimer (in diethyl ether) yet these aggregations have not been identified. And those associated states have not been yet investigated in detail either.
With the computational chemical approach such as molecular orbital calculation, we have already clarified the origin of the conformational characteristics of the methyl group between the isolated MeLi and MeLi crystal which was experimentally observed before. And in this study, considering those results, we try to determine the MeLi associated states in organic solution by means of FEG methodology via molecular dynamics simulation in order to systematically understand the geometrical information (orientation/ aggregation) on the isolated MeLi, MeLi crystal and MeLi in solution, and, at the same time, to reach a mutual understanding between the theory and the experiment.
[Reference]
Y. Ohta, A. Demura, T. Okamoto, H. Hitomi and M. Nagaoka, J. Phys. Chem. B, 110, 12640 (2006).
neutral molecule
分子がプラスにもマイナスにも帯電していない中間の状態のことである。例えば、水分子や酸素分子、アンモニア分子などが挙げられる。ここでいう中性とは酸性、塩基性に対する中性のことではない。
ion
電荷をもつ原子または原子の集団のことである。正、負の電荷をもつものをそれぞれ陽イオン(カチオン)、陰イオン(アニオン)といい、電解質溶液中などには電気的に等量の陽イオンと陰イオンが含まれている。気相や固相中にもイオンは存在する。
stable state
等温かつ定圧の条件下において、運動形式間で分子が変化しうる構造において、最も自由エネルギーの低い状態のことである。分子がとりうる構造の自由エネルギー面を山と谷で表すとすると、谷底の部分のことを指す。
transition state
化学反応の過程で原系から生成系に変換するときに通る最も自由エネルギーの高い状態のことである。例えば、2つの分子の衝突によって反応が開始するとき、力学的エネルギーが分子内部のエネルギーに変換され、元の構造からゆがんだ構造をとるが、その中で最も自由エネルギーの高い状態のことを指す。
molecular orbital
In molecular orbital (MO) calculation, each electronic structure which consists of the molecular is described by MO, i.e., one electron space orbital function extended overall molecular. MO method enables us to evaluate the electron state of molecular via MO calculation.
ammonia
アンモニアは分子式 NH3 で表される無機化合物である。常温常圧では無色で刺激臭のある気体であり、加圧により容易に液化し、水への溶解度が非常に高いといった特異な性質を持つ分子である。化学工業では最も基礎的な窒素源としてきわめて重要である。
methyllithium (CH3Li)
Methyllithium (MeLi) is one of the organometallic reagents, i.e., alkyllithiums (RLis) (R: alkyl group), which have the C-Li bond. It is usually synthesized from the reactions with alkyl halide and metallic lithium. It also forms aggregations in solution, and then, greatly influences on reactivity since its degree decreases by adding the coordination compound.
nucleophilic reagent
Nucleophilic reagent reacts toward the atom of low electron density, and makes some chemical bonds in many cases. Some organometallic reagents, e.g., Grinard reagents/ organolithium compounds, show high reactivity for various substrates, thus, nucleophilic reagent is quite important when the C-C bond needs to be obtained in organic synthesis.
body centered cubic lattice
Body-centered cubic lattice is one of the crystal structures. Atoms are located at the center and vertexes of each unit cell in cubic. Abbreviation: BCC; packing ratio: 68% (not close-packing), coordination number: 8; total number of atom: 2. This type of structure is found in a lot of alkali metals.
Staggered and eclipsed conformation
Suppose the steric conformation of A-B surrounding in X-A-B-Y bond. This can be described as the dihedral angle between X-A and B-Y bond d(X-A-B-Y). If d(X-A-B-Y)= 60, 180 degrees, the substituent groups on A and B exist alternately, and this is called the staggered conformation. On the other hand, if d(X-A-B-Y)= 0, 120 degrees, the substituent groups overlap each other, and this is called the eclipsed one.
free energy
熱力学における状態量の1つ。温度・体積一定の過程における自由エネルギーをヘルムホルツの自由エネルギーF(またはA)、温度・圧力一定の過程における自由エネルギーをギブズの自由エネルギーGと呼ぶ。
free energy gradient method
溶液中での溶質分子の構造最適化手法の一つ。自由エネルギー面上における溶質分子が受ける力、すなわち負の勾配から、溶質構造を更新する。同様の操作を繰り返し行い、ゼロ勾配となったときの構造を最適化構造とする。反応座標に沿って溶質分子の構造最適化を実行することで、自由エネルギー面上の極値である反応系と生成系とを結ぶ自由エネルギープロファイルを求めることが出来る。
glycine
グリシンとはアミノ酢酸のことである。分子式はNH2CH2COOHである。タンパク質を構成するアミノ酸の中で最も単純な形を有する。不斉炭素を持たないために、生体を構成するアミノ酸としては唯一 D-, L- の立体異性が存在しない。
molecular dynamics
2体またはそれ以上の(質点と近似された)原子間にポテンシャルを仮定し、ニュートンの運動方程式を数値的に解くことにより、系全体の運動を解析する方法。電子や核の量子効果を取り入れた手法もある。AMBER、CHARMm、NAMD、GROMACSなどの汎用ソフトウェアがある。
Division of Energy Materials, Institute of Materials Innovation,
Institute of Innovation for Future Society, Nagoya University
F3-4 Furo-cho, Chikusa-ku, Nagoya 464-8601, JAPAN